Panorama general de la FMD de la UE
Con la Directiva de Medicamentos Falsificados (FMD) publicada en julio de 2011, la Agencia Europea de Medicamentos (EMA) establecer el comienzo de los estándares de trazabilidad de los países de la Unión Europea.
El Directiva sobre medicamentos falsificados (Directiva 2011/62/UE) introduce medidas europeas armonizadas para luchar contra las falsificaciones de medicamentos y garantizar que los medicamentos sean seguros y que los medicamentos en circulación comercial estén estrictamente controlados. Las medidas incluyen:
- Elementos de seguridad obligatorios (un identificador único y un dispositivo antimanipulación) en el embalaje exterior de los medicamentos
- Un logotipo común en toda la UE para identificar las farmacias en línea legales
- Normas más estrictas sobre la importación de principios activos farmacéuticos
- Requisitos de mantenimiento de registros reforzados para distribuidores mayoristas
A partir de 2019, todos los productos farmacéuticos deben cumplir plenamente las obligaciones de la fiebre aftosa. Hasta 2025, los países de la UE con un sistema separado, como Grecia e Italia, deben cumplir plenamente con la regulación de seguimiento y localización farmacéutica.
Requisitos de serialización
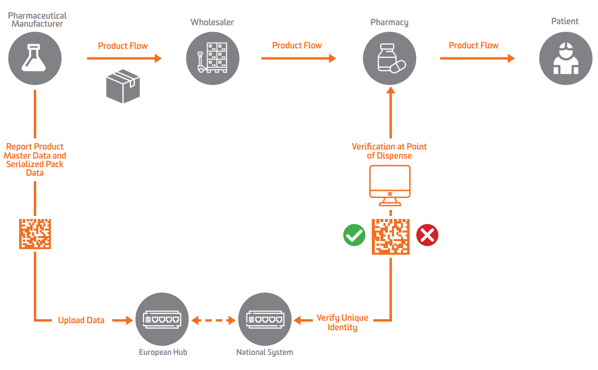
De acuerdo con la FMD de la UE, la serialización debe aparecer en el nivel secundario o de unidad vendible en Europa. Para habilitar la verificación, los fabricantes primero deben serializar el producto y enviar esos datos serializados a un repositorio central que pueda realizar consultas en su contra.
Para permitir la serialización, verificación y presentación de informes a las autoridades, EU FMD exige que los fabricantes marquen los paquetes con cuatro elementos de datos que deben imprimirse en un formato legible por humanos y codificarse y almacenarse en un GS1 2D DataMatrix:
- Identificador de Producto
- Número de serie
- Número de lote o lote
- Fecha de caducidad
El uso del quinto elemento de datos, el número de reembolso nacional, es opcional, y pocos estados de la UE pueden solicitar incluirlo en un identificador único para vincular el reembolso de un medicamento en el marco de un programa de medicina socializada.
Los fabricantes de productos farmacéuticos y los comerciantes paralelos deben informar datos a un Hub central de la UE dirigido por el Organización Europea de Verificación de Medicamentos (EMVO), que también incorpora a los usuarios al Hub. Esto enviará los datos a los repositorios de datos apropiados administrados por las Organizaciones Nacionales de Verificación de Medicamentos (NMVO) correspondientes, que son responsables de la incorporación de usuarios finales y del funcionamiento de los sistemas nacionales.
Los requisitos de información
Según la FMD de la UE, el titular de la autorización de comercialización (TAC) debe enviar los datos maestros del producto y los datos del paquete del producto serializado.
Los datos maestros incluyen:
- Códigos de producto
- Forma
- Fortaleza
- Dosis por paquete
- Tipo de paquete
- Mercado(s) objetivo para la distribución
- Futuro y cualquier actualización de los datos maestros del producto
Los datos del paquete de productos serializados incluyen:
- Códigos de producto
- Número de lote/lote
- Fecha de caducidad
- Números seriales
- Cualquier actualización de los datos del paquete de productos serializados