نظرة عامة على مرض الحمى القلاعية في الاتحاد الأوروبي
مع توجيه الأدوية المغشوشة (FMD) المنشور في يوليو 2011 ، فإن وكالة الأدوية الأوروبية (EMA) وضع بداية معايير التتبع لدول الاتحاد الأوروبي.
ال التوجيه بشأن الأدوية المغشوشة (التوجيه 2011/62 / EU) يقدم تدابير أوروبية منسقة لمكافحة تزوير الأدوية والتأكد من أن الأدوية آمنة وأن الأدوية المتداولة في التجارة تخضع لرقابة صارمة. التدابير تشمل:
- ميزات أمان إلزامية - معرّف فريد وجهاز مضاد للعبث - على العبوة الخارجية للأدوية
- شعار شائع على مستوى الاتحاد الأوروبي لتحديد صيدليات الإنترنت القانونية
- قواعد أكثر صرامة على استيراد المكونات الصيدلانية الفعالة
- تعزيز متطلبات حفظ السجلات لموزعي الجملة
اعتبارًا من عام 2019 ، يجب أن تفي جميع المنتجات الصيدلانية تمامًا بالتزامات مرض الحمى القلاعية. حتى عام 2025 ، يجب أن تكون دول الاتحاد الأوروبي التي لديها نظام منفصل مثل اليونان وإيطاليا متوافقة تمامًا مع المسار الصيدلاني وتنظيم التتبع.
متطلبات التسلسل
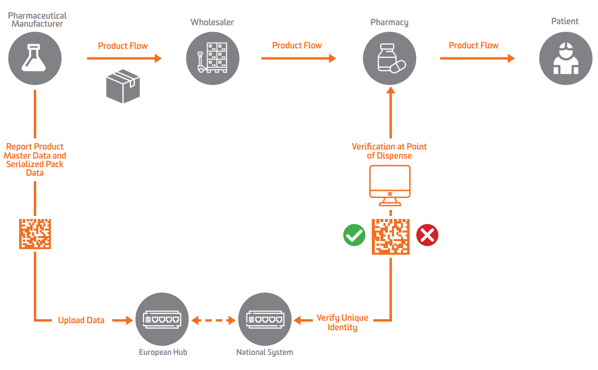
وفقًا لـ EU FMD ، يجب أن تظهر التسلسل على مستوى الوحدة الثانوية أو القابلة للبيع في أوروبا. لتمكين التحقق ، يحتاج المصنعون إلى إجراء تسلسل للمنتج أولاً وإرسال تلك البيانات المتسلسلة إلى مستودع مركزي يمكنه إجراء استعلامات ضده.
لتمكين التسلسل والتحقق والإبلاغ إلى السلطات، يتطلب الاتحاد الأوروبي FMD أن تقوم الشركات المصنعة بوضع علامة على الحزم بأربعة عناصر بيانات يجب طباعتها في شكل يمكن قراءته بواسطة الإنسان، وترميزها وتخزينها في GS1 2D DataMatrix:
- معرف المنتج
- رقم سري
- رقم الدفعة أو الدفعة
- تاريخ الانتهاء
يعد استخدام عنصر البيانات الخامس ــ رقم السداد الوطني ــ اختياريا، وقد تطلب دول قليلة في الاتحاد الأوروبي إدراج نفس العنصر في معرف فريد لربط سداد تكاليف منتج دوائي في إطار برنامج الطب الاجتماعي.
يجب على مصنعي الأدوية والتجار الموازيين إبلاغ البيانات إلى مركز مركزي في الاتحاد الأوروبي يديرها المنظمة الأوروبية للتحقق من الأدوية (EMVO)، والتي تعمل أيضًا على ضم المستخدمين إلى Hub. سيؤدي ذلك إلى دفع البيانات إلى مستودعات البيانات المناسبة التي تديرها المنظمات الوطنية المعنية للتحقق من الأدوية (NMVOs) ، المسؤولة عن ضم المستخدم النهائي وتشغيل الأنظمة الوطنية.
متطلبات تقديم التقارير
بموجب الاتحاد الأوروبي FMD ، يتعين على صاحب ترخيص التسويق (MAH) تقديم البيانات الرئيسية للمنتج وبيانات حزمة المنتج التسلسلية.
تتضمن البيانات الرئيسية:
- أكواد المنتج
- استمارة
- قوة
- جرعات لكل علبة
- نوع العبوة
- السوق (الأسواق) المستهدفة للتوزيع
- المستقبل وأي تحديثات للبيانات الرئيسية للمنتج
تتضمن بيانات حزمة المنتج المسلسلة ما يلي:
- أكواد المنتج
- رقم الدفعة / الدفعة
- تاريخ الانتهاء
- الأرقام التسلسلية
- أي تحديثات لبيانات حزمة المنتج المتسلسلة
اكتشف كيف يمكن أن تساعدك SoftGroup في تحقيق الامتثال للاتحاد الأوروبي بشأن مرض الحمى القلاعية
عرض الحلول